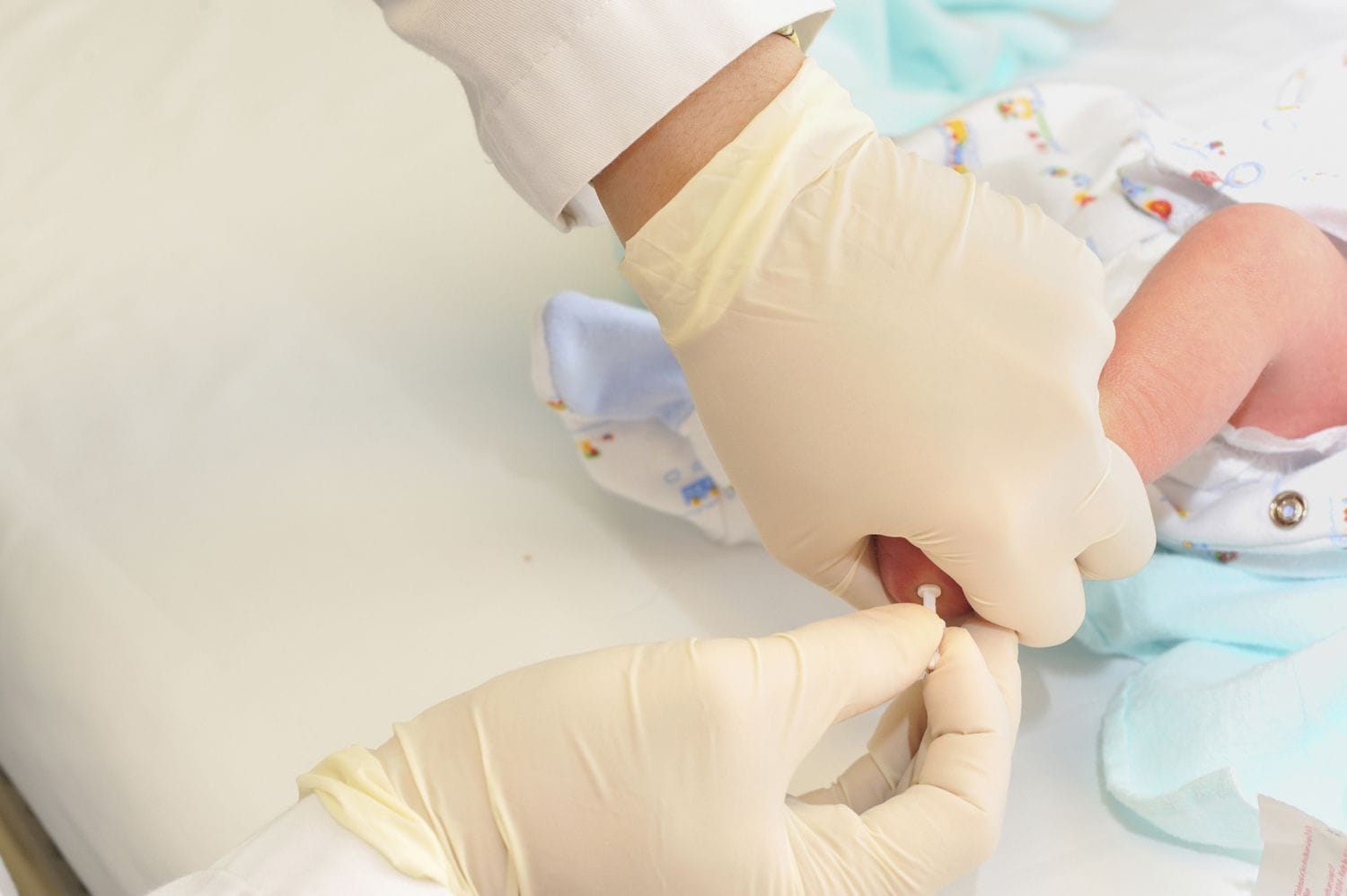
Phenylketonuria (PKU) is a metabolic disease caused by a genetic mutation. This disease used to be very difficult to diagnose, but for the last 40+ years, a PKU test has been a part of the neonatal screening process in the United States.
If not immediately, continually, and properly treated by a particular diet, PKU can result in severe mental disability, heart problems, seizures, and other physical, skin-related, and neurological issues.
What is PKU and what causes it?
Phenylketonuria (PKU) is a rare, autosomal recessive disease that prevents the body from breaking down one of the amino acids found in nearly all proteins: phenylalanine (Phe). The gene affected, PAH, encodes phenylalanine hydroxylase, which converts phenylalanine to tyrosine (another amino acid) in the liver. When this conversion does not happen, phenylalanine builds up in the blood and causes problems, particularly in the brain.
It occurs when an abnormal or mutated PAH gene from both the mother and the father is passed on to the baby. Each person has two copies of this gene, one from each parent. Two mutated or deleted PAH genes causes PKU. This can mean one of four things:
- The parents are both carriers of 1 copy of the mutated gene each (but do not have the disease)
- One parent has the disease and the other is a carrier of a mutated gene
- Both parents have the disease, or
- One parent is a carrier of a mutated gene and the other abnormal gene came about through a mutation or deletion of the gene of the other’s egg or sperm.
There are a few other common names for Phenylketonuria (PKU) to be aware of:
- Folling’s disease
- Phenylalanine hydroxylase deficiency
- PAH deficiency
How common is PKU in infants?
Between 1 in 10,000 to 15,000 newborns are affected in the USA. It is considered a relatively rare disease.
If you and your partner’s families have a history of PKU, you may want to consider genetic counseling to understand further your chances of conceiving a child with the condition.
Typically, if both parents are carriers of the mutated gene (each has one mutated copy), then only 25% of each pregnancy is at risk to have the full disease. This also means that 50% of each pregnancy is likely to result in a child who is also a carrier of the mutated gene.
What are the symptoms of PKU?
Typically, a newborn with PKU is identified in the newborn screening and starts treatment immediately, meaning that it is unlikely even to notice symptoms. If it is not caught in the newborn screen, though highly unlikely, by the time it is detectable through symptoms the brain damage is often irreversible.
If not diagnosed and placed on a PKU diet soon after birth, the following symptoms may be detected:
- Lethargy (weakness)
- Poor feeding habits
- Vomiting
- Irritability
- Skin rash with blisters/pimples (eczema-like)
- Musty (“mouse-like”) body odor
If not kept on the PKU diet, the following are the most common neurological symptoms:
- Seizures
- Spastic muscle movements
- Hypertonicity (tight muscles)
- Increased tendon reflexes
- Abnormal EEG
- Physical disability (rare)
How does PKU cause brain damage or mental disability?
If untreated or poorly regulated, the high levels of Phe in the blood build up in the brain. These elevated levels of Phe cause the destruction of the fatty insulating layer called myelin that surrounds nerve fibers of the brain. Without myelin, the nerves cannot fire or communicate correctly, resulting in mental retardation.
How is PKU diagnosed?
PKU is diagnosed through a routine neonatal screening performed at the hospital by law in the USA, as well as in many other developed countries. The test is performed as soon as the child is born and involves taking a drop of blood from the baby’s heel. This way, the healthcare staff, and the child’s parents can provide adequate treatment to prevent brain damage.
What kind of treatments are available?
The treatment available for PKU consists of a slew of dietary restrictions. This includes limiting protein intake; however, this must be done cautiously to prevent mental retardation, as well as neurological and dermatological problems. The following are common high-protein/high phenylalanine foods that should be avoided:
- Dairy (milk, cheese, yogurt, etc.)
- Eggs
- Nuts, beans, and legumes
- Soybeans and peas
- Poultry, beef, and pork
- Fish and shellfish
- Beer (later in life)
- The artificial sweetener aspartame
In the United States, it is required by law that if a food contains aspartame (ex. diet sodas), it must include the label:
Phenylketonurics: Contains phenylalanine.
Since the PKU diet doesn’t allow a large amount of protein, people with PKU may lack necessary nutrients. This is why it is important to talk with your doctor and/or a nutritionist to ensure you or your child receive all the necessary nutrients.
KUVAN is a unique formula approved by the FDA to be used when following the PKU treatment. Kuvan is a synthetic form of a co-factor (BH4) that joins with the PAH enzyme to convert phenylalanine to tyrosine. For many PKU patients, Kuvan can help to lower the amount of phenylalanine in the body. It is a daily oral tablet or powder form that can be mixed into a drink.
The same company that developed Kuvan began Phase III clinical trials for a new treatment in 2016, called Pegvaliase, or PEG-PAL. PEG-PAL is a once-daily injection that has been shown to profoundly reduce the amount of phenylalanine in the body, even down to normal levels. Phase III trials did not show any improvement in mental concentration or ability, however. It may be released for use after further talks with the FDA.
Is there a cure for PKU?
Since PKU is a genetic disease, it does not have a cure. The mutations of the two copies of the PAH gene are present in every cell in the affected person’s body. As mentioned above, there are treatments available that can control the circulating amount of phenylalanine in the body and reduce it to a safe level. Keeping a specific low-Phe (low protein) diet can be a very effective treatment as well.
As mentioned above, there are treatments available, such as keeping a specific low-Phe (low protein) diet. There are also medications that can control the circulating amount of phenylalanine in the body and reduce it to a safe level (the examples above are Kuvan and PEG-PAL).
Are there other conditions related to PKU?
There are a few conditions that also can cause an excess of phenylalanine in the blood. The disorder that is most closely related to PKU is called tetrahydrobiopterin deficiency phenylalaninemia, or tetrahydrobiopterin (BH4) deficiency. Persons with this disorder may have perfectly healthy PAH genes, and instead have mutations in the gene that encodes BH4, a coenzyme that binds with phenylalanine hydroxylase to help convert phenylalanine to tyrosine. It is also active with other enzymes and, along with high Phe levels, can result in low neurotransmitter levels. This can cause neurological problems similar to those seen with PKU.
This condition is also infrequent and requires immediate treatment to prevent long-term neurological and movement issues.
Mothers with PKU:
I have PKU, and I want to have a baby/am already pregnant. Are there any risks for my baby?
The greatest risks come if a woman is untreated for PKU/does not follow a PKU diet and/or take Phe-reducing medications. If a woman goes untreated, she can present problems in pregnancies such as an increase in spontaneous miscarriages or delayed fetal growth. Additionally, pregnant women who have untreated PKU are more likely to have children with microcephaly, congenital heart disease, and facial abnormalities. The association with these issues is greater at higher maternal levels of phenylalanine.
The important thing to remember is that the association with these issues is more significant at higher maternal levels of phenylalanine. Therefore, if you have PKU and are in a treatment regimen and have controlled serum levels of Phe, your chances of having a baby with the issues above are considerably lower. If you are concerned, take the time to speak with your doctor about steps to take before you conceive, or things you may need to focus on now that you are pregnant.
Doctors recommend that if a woman with PKU wants to conceive and is not currently on a PKU-restricted diet or Phe-reducing medications, that she begin these regimens before conceiving.
If I have a baby with PKU, can he or she breastfeed?
Since breastmilk contains a significant amount of protein, and thus phenylalanine, no – it is not considered safe to breastfeed your child. In this case, the risks of breastfeeding outweigh the benefits. The doctors will prescribe appropriate formula and medication so that your baby can stay at a safe level of phenylalanine.
If I have PKU, does that mean my baby will too?
This depends completely on the father. If you as the mother have classical PKU (two severely mutated copies of the PAH gene), then you will pass one of these mutated copies onto your baby, making him or her a carrier. If the father does not have it and is not a carrier of a mutated gene copy, then there is nearly no chance of your baby having classical PKU (the “nearly no” refers to the highly unlikely possibility of a sperm having a random mutation in that gene). If the father is a carrier of a mutated PAH gene, then there is a 50% chance that your baby will have PKU, and a 50% chance that the baby will only be a carrier.
If you have PKU and you know that your partner is a carrier, you may want to speak with a genetic counselor prior to trying to conceive.
Compiled using information from the following sources:
1. National Organization for Rare Disorders (NORD): Phenylketonuria.
2. US National Library of Medicine: Genetics Home Reference: Phenylketonuria.
3. National Institute of Child Health and Human Development: Phenylketonuria (PKU).
https://nichd.nih.gov/health/topics/pku/conditioninfo/Pages/treatments.aspx
4. National PKU Alliance: Education: About PKU.
https://www.npkua.org/Education/About-PKU
5. ClinicalTrials.gov: Phase 3 Study to Evaluate the Efficacy & Safety of Self-Administered Injections of BMN165 by Adults With PKU.
https://clinicaltrials.gov/ct2/show/NCT01889862
6. Rare Disease Report (RareDR): PKU News: BioMarin Submits Peg-Pal Application to the FDA.
Want to Know More?
- Genetic Counseling
- Progeria: Hutchinson-Gilford Progeria Syndrome (HGPS)



